
Cancer type > Hematological Cancers
Definition of Wilms tumor
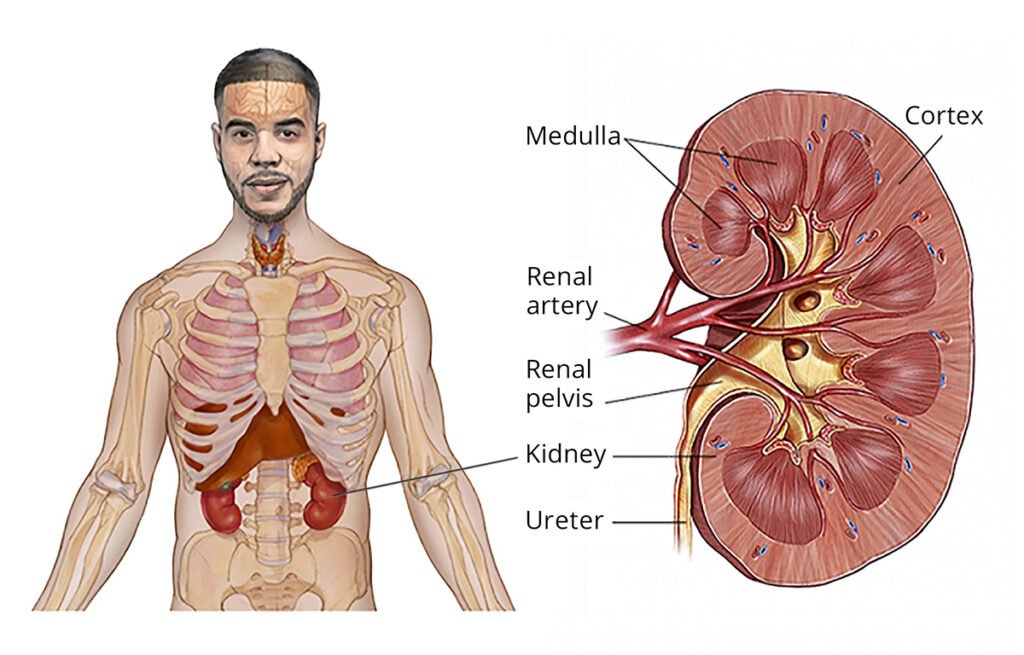
Wilms tumor, also known as nephroblastoma, is a rare type of kidney cancer that primarily affects children. It is the most common malignant kidney tumor in childhood, typically occurring in children under the age of 5.
Key Characteristics
- Origin:
- Wilms tumor arises from embryonic kidney cells (metanephric blastema), which are involved in kidney development during fetal growth.
- The tumor often contains a mix of tissues, including epithelial, stromal, and blastemal components.
- Unilateral vs. Bilateral:
- In most cases, it affects only one kidney (unilateral).
- In rare cases, it can develop in both kidneys (bilateral).
- Age Group:
- The majority of cases are diagnosed between 3 to 4 years old and are uncommon after the age of 6.
- Prognosis:
- With early detection and modern treatments, the prognosis for Wilms tumor is generally excellent.
Clinical Features
Children with Wilms tumor may present with:
- A painless abdominal mass or swelling (most common sign).
- Fever, hematuria (blood in urine), hypertension (due to kidney involvement).
- Loss of appetite, nausea, or weight loss in advanced cases.
Wilms tumor is a highly treatable condition, especially when detected early, and is managed with a combination of surgery, chemotherapy, and, in some cases, radiation therapy.
The symptoms of Wilms tumor can vary depending on the size and spread of the tumor. Many children with Wilms tumor are otherwise healthy, and the condition is often detected incidentally during a routine check-up or when parents notice an unusual abdominal swelling.
SYMPTOMS OF WILMS TUMOUR
- Abdominal Mass or Swelling:
- The most frequent and noticeable symptom.
- Often painless and firm to touch.
- May be confined to one side of the abdomen.
- Abdominal Pain:
- May occur as the tumor grows larger and presses on nearby organs.
- Hematuria (Blood in Urine):
- Blood may be visible in the urine, or detected through microscopic examination.
- Fever:
- Often unexplained and low-grade.
- Hypertension (High Blood Pressure):
- Caused by pressure on the kidney or hormones released by the tumor.
Other Symptoms
- Loss of Appetite or nausea.
- Weight Loss.
- Fatigue or decreased energy levels.
- Constipation (if the tumor presses on the intestines).
- Shortness of Breath (in rare cases, if the tumor spreads to the lungs).
Symptoms of Advanced Disease
If the tumor has metastasized (spread), additional symptoms may include:
- Respiratory Issues: Due to lung involvement (e.g., cough, difficulty breathing).
- Bone Pain: From metastases in bones.
- Swelling in Limbs: If the tumor compresses blood vessels.
When to Seek Medical Attention
Parents should consult a healthcare provider if they notice:
- A firm, swollen abdomen in a child.
- Persistent abdominal discomfort or other unexplained symptoms.
Early diagnosis and treatment are crucial for favorable outcomes in children with Wilms tumor.
TYPES OF WILMS TUMOUR
Wilms tumor can be categorized based on histology (cell appearance under a microscope) and genetic or syndromic associations. These classifications are crucial for determining the treatment approach and prognosis.
- Types Based on Histology
- Favorable Histology
- Description:
- The most common type, accounting for about 90% of cases.
- Tumor cells look relatively normal and lack significant abnormalities.
- There is no anaplasia (abnormal cell growth and division).
- Prognosis:
- Excellent with treatment.
- Unfavorable Histology (Anaplastic Wilms Tumor)
- Description:
- Tumor cells show anaplasia, characterized by:
- Abnormal nuclei.
- Resistance to therapy.
- Two forms:
- Focal anaplasia: Limited to specific areas of the tumor.
- Diffuse anaplasia: Widespread throughout the tumor.
- Tumor cells show anaplasia, characterized by:
- Prognosis:
- Poorer than favorable histology, particularly in cases of diffuse anaplasia.
- Requires more aggressive treatment.
- Types Based on Syndromic Associations
Wilms tumor is sometimes associated with congenital syndromes or genetic abnormalities:
- Sporadic Wilms Tumor
- Description:
- Occurs without any known genetic or syndromic predisposition.
- Most common presentation.
- Syndromic Wilms Tumor
- Description:
- Associated with specific syndromes or genetic conditions that predispose to tumor development.
- Examples:
- WAGR Syndrome:
- Includes Wilms tumor, Aniridia (absence of the iris), Genitourinary anomalies, and Retardation (intellectual disability).
- Associated with deletion on chromosome 11 (WT1 gene).
- Beckwith-Wiedemann Syndrome (BWS):
- Characterized by overgrowth, macroglossia (large tongue), and abdominal wall defects.
- Associated with abnormalities in chromosome 11 (IGF2 gene).
- Denys-Drash Syndrome:
- Includes Wilms tumor, kidney disease, and ambiguous genitalia.
- Caused by mutations in the WT1 gene.
- Perlman Syndrome:
- Rare, with features like overgrowth and an increased risk of Wilms tumor.
- WAGR Syndrome:
- Genetic and Molecular Subtypes
Wilms tumor can also be classified based on molecular or genetic features, which influence prognosis:
- WT1 Mutation-Associated Tumors:
- Seen in syndromic and some sporadic cases.
- WT2 Abnormalities:
- Common in Beckwith-Wiedemann syndrome.
- Loss of Heterozygosity (LOH):
- Loss of regions on chromosomes 1p and 16q is associated with poorer outcomes.
- Gain of Chromosome 1q:
- Linked to a higher risk of relapse.
Understanding the specific type of Wilms tumor helps guide treatment planning and provides insights into prognosis and potential familial implications.
The exact cause of Wilms tumor (nephroblastoma) is not fully understood, but it is believed to result from abnormalities in the development of kidney cells during fetal growth. These abnormalities may cause cells that should mature into kidney cells to remain immature, leading to uncontrolled cell growth.
Causes and Risk factors Wilms Tumor
- Genetic Mutations:
- Mutations in specific genes that regulate cell growth and development are implicated in Wilms tumor. Key genes include:
- WT1 (Wilms Tumor 1): Plays a crucial role in kidney and genitourinary system development.
- WT2: Found on chromosome 11, abnormalities are linked to syndromes associated with Wilms tumor.
- IGF2: Overexpression may contribute to tumor development, especially in Beckwith-Wiedemann Syndrome.
- Mutations in specific genes that regulate cell growth and development are implicated in Wilms tumor. Key genes include:
- Developmental Errors:
- Wilms tumor originates from remnants of embryonic kidney cells (metanephric blastema) that fail to mature properly.
- Epigenetic Changes:
- Changes in gene regulation without alterations in the DNA sequence may also play a role.
Risk Factors
- Age
- Most cases occur in children aged 3 to 4 years.
- Rarely diagnosed after the age of 6.
- Gender
- Slightly more common in girls than in boys.
- Race/Ethnicity
- More common in African American children.
- Less common in Asian populations.
- Family History
- A family history of Wilms tumor increases the risk.
- Inherited cases are rare (1–2%) but may follow an autosomal dominant inheritance pattern.
- Congenital Syndromes
Certain syndromes are strongly associated with Wilms tumor, often due to genetic mutations:
- WAGR Syndrome:
- Deletion of part of chromosome 11 (WT1 gene).
- Features include Wilms tumor, aniridia (absence of the iris), genitourinary anomalies, and developmental delays.
- Beckwith-Wiedemann Syndrome (BWS):
- Overgrowth disorder associated with mutations in WT2 and IGF2 genes.
- Other features include macroglossia (large tongue) and abdominal wall defects.
- Denys-Drash Syndrome:
- Caused by WT1 mutations.
- Includes kidney disease, ambiguous genitalia, and a high risk of Wilms tumor.
- Perlman Syndrome:
- Rare overgrowth syndrome with a high predisposition to Wilms tumor.
- Genetic Abnormalities
- Loss of Heterozygosity (LOH):
- LOH at chromosomes 1p and 16q is linked to a higher risk of developing Wilms tumor and poorer prognosis.
- Gain of 1q:
- Associated with worse outcomes.
- Birth Defects
- Certain congenital anomalies increase the risk of Wilms tumor, such as:
- Hypospadias (urethral opening on the underside of the penis).
- Cryptorchidism (undescended testicle).
- Hemihypertrophy (one side of the body larger than the other).
Are There Preventable Risk Factors?
- Most cases of Wilms tumor are not preventable, as they are associated with genetic mutations and developmental abnormalities that occur during fetal growth.
- There is no evidence that parental behaviors or environmental factors during pregnancy significantly contribute to the development of Wilms tumor.
Early recognition of symptoms and awareness of associated risk factors (such as congenital anomalies or syndromes) are critical for timely diagnosis and treatment. Genetic counseling may benefit families with a history of Wilms tumor or associated syndromes.
TREATMENT OF WILMS TUMOR
The treatment of Wilms tumor is based on factors such as the tumor stage, histology (favorable or unfavorable), the child’s age, and overall health. It typically involves a combination of surgery, chemotherapy, and radiation therapy, with a multidisciplinary team approach.
- Surgery
- Nephrectomy (removal of the affected kidney) is the primary surgical treatment.
- Radical Nephrectomy: Removes the kidney, adrenal gland, and surrounding tissues.
- Partial Nephrectomy: Removes only the tumor and part of the kidney, used for children with bilateral tumors or at risk of kidney failure.
- Surgery helps:
- Diagnose and stage the tumor.
- Remove the bulk of the cancer.
- For advanced or large tumors, surgery may follow chemotherapy to shrink the tumor.
- Chemotherapy
- Used in most cases of Wilms tumor, either before or after surgery.
- Commonly Used Drugs:
- Actinomycin D (Dactinomycin)
- Vincristine
- Doxorubicin (used for more advanced stages or unfavorable histology).
- Purpose:
- Pre-surgical chemotherapy may reduce tumor size.
- Post-surgical chemotherapy treats remaining cancer cells and prevents recurrence.
- Radiation Therapy
- Typically used for:
- Advanced stages of the disease.
- Unfavorable histology (e.g., anaplastic Wilms tumor).
- Tumors that cannot be completely removed by surgery.
- Targets the abdomen or areas where cancer has spread (e.g., lungs or liver).
- Treatment by Stage
Stage I (Confined to the Kidney)
- Surgery (nephrectomy).
- Chemotherapy (actinomycin D and vincristine).
- No radiation therapy for favorable histology.
Stage II (Spread to Nearby Tissues)
- Surgery.
- Chemotherapy (actinomycin D and vincristine).
- Radiation therapy if histology is unfavorable.
Stage III (Spread to Lymph Nodes or Other Structures in the Abdomen)
- Surgery.
- Chemotherapy (actinomycin D, vincristine, and doxorubicin).
- Radiation therapy to the abdomen.
Stage IV (Spread to Distant Organs like Lungs, Liver, or Bones)
- Surgery.
- Chemotherapy (intensive regimen including doxorubicin).
- Radiation therapy to metastatic sites.
Stage V (Both Kidneys Involved)
- Chemotherapy to shrink tumors in both kidneys.
- Partial nephrectomy to preserve kidney function.
- Additional chemotherapy and possible radiation therapy.
- Stem Cell Transplantation
- Used in rare cases of relapsed or high-risk Wilms tumor.
- High-dose chemotherapy is followed by stem cell transplantation to restore bone marrow function.
- Targeted and Experimental Therapies
- Targeted Therapy: Investigational drugs targeting specific genetic mutations (e.g., WT1, IGF2) are being studied.
- Clinical Trials: Families may consider enrolling in trials for access to cutting-edge treatments.
- Supportive Care
- Pain management, nutrition, and psychosocial support are essential.
- Management of treatment side effects like nausea, fatigue, or hair loss.
Prognosis
- Prognosis is generally excellent for children with favorable histology, with a survival rate of over 90% for early-stage disease.
- Outcomes are more challenging for those with unfavorable histology or advanced stages, but modern treatments continue to improve survival rates.
Treatment is highly individualized, and a pediatric oncologist will guide the family through the most appropriate options. Regular follow-ups are essential to monitor for recurrence or late side effects of treatment.
Currently, there is no known way to prevent Wilms tumor, as it typically arises from genetic and developmental abnormalities in kidney cells during fetal growth. These factors are not influenced by modifiable lifestyle or environmental exposures.
Why Prevention Is Challenging
- Genetic Origins:
- Wilms tumor often results from mutations in specific genes (WT1, WT2, or IGF2) or errors during embryonic kidney development, which occur randomly.
- These genetic abnormalities are not preventable.
- Congenital Syndromes:
- Inherited conditions like WAGR syndrome, Beckwith-Wiedemann syndrome, and Denys-Drash syndrome predispose to Wilms tumor. These are rare and related to genetic abnormalities present at birth.
- Non-Modifiable Risk Factors:
- Most risk factors for Wilms tumor (e.g., age, race, family history, congenital anomalies) are beyond control.
PREVENTION OF WILMS TUMOR
While prevention is not possible, families with an elevated risk due to genetic factors or syndromes can take steps for early detection, which significantly improves outcomes.
- Genetic Counseling
- Families with a history of Wilms tumor or associated syndromes should seek genetic counseling.
- Testing for mutations in genes like WT1 or WT2 can identify children at higher risk.
- Regular Screening for High-Risk Groups
- Children with syndromes associated with Wilms tumor (e.g., Beckwith-Wiedemann, WAGR) should undergo routine abdominal ultrasounds and physical exams to detect tumors early.
- Screening may be recommended every 3-4 months until the child reaches age 8.
- Prenatal and Pediatric Monitoring
- Parents should ensure regular pediatric check-ups.
- Unusual symptoms, such as abdominal swelling or persistent pain, should be evaluated promptly.
Healthy Lifestyle During Pregnancy
Although there is no evidence linking maternal behavior or environmental exposures directly to Wilms tumor, maintaining overall health during pregnancy may reduce the risk of other complications:
- Avoid tobacco, alcohol, and harmful substances.
- Eat a balanced diet and follow prenatal care guidelines.
Research Directions
Ongoing studies aim to identify genetic or molecular triggers for Wilms tumor. Future advancements in understanding these mechanisms may lead to:
- Preventive interventions for high-risk populations.
- Early gene-based detection techniques.
For now, focus remains on early diagnosis, effective treatment, and genetic counseling for at-risk families. Early detection has proven critical in achieving high survival rates for children with Wilms tumor.
NEWER ADVANCEMENTS AND RESEARCHES
Recent advancements in the understanding and treatment of Wilms tumor have led to improved outcomes and more personalized therapeutic approaches. Key areas of progress include:
- Genetic and Molecular Insights
- Identification of Genetic Mutations: Research has uncovered specific genetic mutations associated with Wilms tumor, such as alterations in the WT1 and WT2 genes. These discoveries enhance diagnostic accuracy and enable the development of targeted therapies.
- Risk Stratification Models: Advanced machine learning techniques have been employed to construct risk models for Wilms tumor, incorporating genetic and clinical data to predict patient outcomes more accurately.
- Treatment Innovations
- Nephron-Sparing Surgery: For children with bilateral Wilms tumor or those at risk of kidney failure, nephron-sparing surgery aims to remove the tumor while preserving as much healthy kidney tissue as possible. This approach reduces the long-term risk of renal insufficiency.
- Targeted Therapies: Investigations into therapies that specifically target genetic mutations found in Wilms tumor cells are ongoing. These treatments aim to minimize damage to healthy cells and reduce side effects.
- Surveillance and Follow-Up
- Updated Surveillance Guidelines: New guidelines have been established for monitoring patients with predisposition syndromes associated with Wilms tumor. These guidelines recommend regular imaging and clinical evaluations to detect tumors at an early, more treatable stage.
- International Collaboration
- Global Clinical Trials: Collaborative efforts among international research groups have led to large-scale clinical trials, facilitating the development of standardized treatment protocols and the sharing of data to improve patient outcomes worldwide.
These advancements reflect a comprehensive approach to improving the diagnosis, treatment, and long-term care of children with Wilms tumor, emphasizing personalized medicine and collaborative research efforts.