
Cancer type > Hematological Cancers
Definition of Neuroblastoma
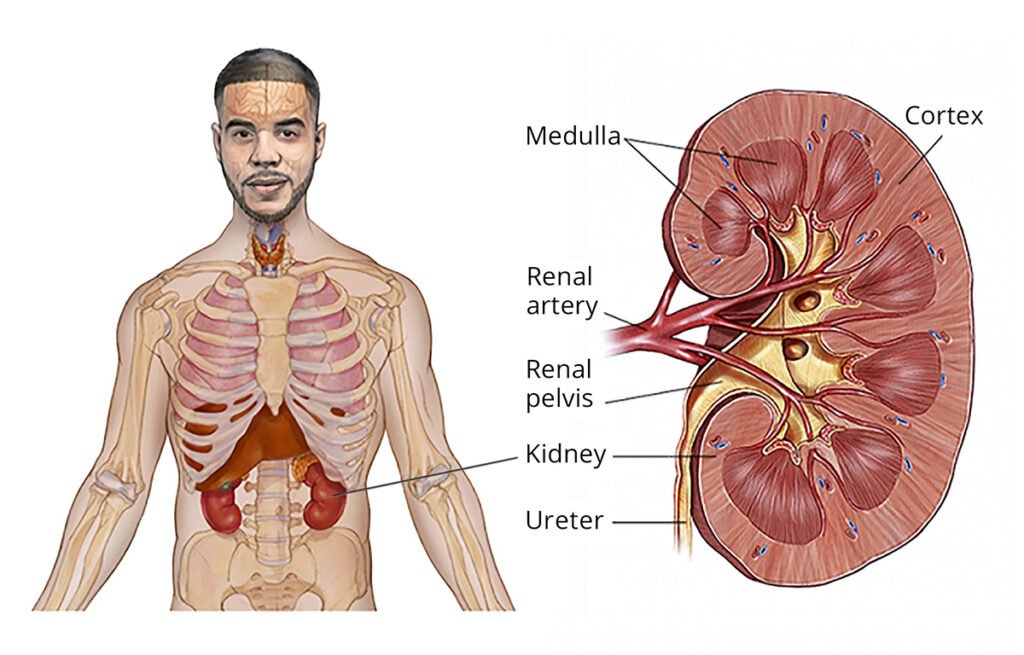
Neuroblastoma is a rare and aggressive type of cancer that develops from immature nerve cells, called neuroblasts, which are part of the sympathetic nervous system. It most commonly arises in the adrenal glands (located on top of the kidneys) but can also occur in nerve tissue along the spine, chest, abdomen, or pelvis.
This cancer primarily affects infants and young children, typically under the age of 5, and is rarely diagnosed in older children or adults. Neuroblastoma can vary widely in its behavior, ranging from cases that spontaneously regress without treatment to aggressive forms that spread (metastasize) to other parts of the body, such as the bone marrow, bones, liver, or lymph nodes.
Key Features:
Origin: Develops from neural crest cells, which are involved in the development of the sympathetic nervous system.
Age Group: Most commonly affects children under 5 years old.
Symptoms: Symptoms depend on the tumor’s location and extent, often including abdominal pain, swelling, or fatigue.
Prognosis: Can range from favorable outcomes in localized cases to a more challenging prognosis in advanced-stage disease.
Neuroblastoma is a type of cancer that develops from immature nerve cells, most commonly found in and around the adrenal glands. It can also develop in nerve tissues along the spine, chest, abdomen, or pelvis. Symptoms vary depending on the location and extent of the tumor but often include:
General Symptoms
Fatigue or weakness
Loss of appetite
Unexplained weight loss
Fever
Localized Symptoms
Abdomen (most common location):
Abdominal pain
Swelling or a mass that can be felt
Constipation or difficulty urinating due to pressure on nearby organs
Chest:
Chest pain
Difficulty breathing or wheezing
Cough
Spinal Cord (if tumor presses on the spine):
Weakness or paralysis
Difficulty walking
Bowel or bladder dysfunction
Symptoms Due to Metastasis
Bone pain (if cancer has spread to the bones)
Limping or inability to bear weight
Swelling or bruising around the eyes (“raccoon eyes”)
Skin lumps (blue or purple in color, indicating spread to the skin)
Paraneoplastic Symptoms (caused by hormones secreted by the tumor):
High blood pressure
Rapid heartbeat
Sweating
Diarrhea
Flushing of the skin
Other Possible Signs
Protruding eyes
Drooping eyelids or unequal pupils (Horner syndrome)
Persistent infections due to immune system suppression
If neuroblastoma is suspected, a healthcare professional typically conducts diagnostic tests such as imaging studies (ultrasound, MRI, CT scans), biopsy, and blood or urine tests to confirm the diagnosis. Early detection and treatment are crucial for better outcomes.
Neuroblastoma can be classified into different types based on clinical presentation, genetic and molecular characteristics, and risk stratification. These classifications help guide treatment and prognosis. Below are the main types and categorizations of neuroblastoma:
TYPES OF NEUROBLASTOMA
Clinical Types
Localized Neuroblastoma:
The tumor is confined to one area and has not spread.
May be treated effectively with surgery alone.
Metastatic Neuroblastoma:
The cancer has spread to distant sites such as the bone, liver, lymph nodes, or bone marrow.
Common in high-risk neuroblastoma cases.
Stage 4S (Special Neuroblastoma):
Occurs in infants under 1 year of age.
Tumor has spread to the skin, liver, or bone marrow, but prognosis is often better because it may regress spontaneously.
Histological Types (Based on Cell Appearance)
Differentiating Neuroblastoma:
Tumor cells show signs of maturing into nerve cells.
Often associated with a better prognosis.
Undifferentiated or Poorly Differentiated Neuroblastoma:
Cells lack features of normal nerve cells.
Associated with a more aggressive course.
Ganglioneuroblastoma:
Contains a mix of mature ganglion cells and immature neuroblast cells.
Can behave less aggressively than pure neuroblastoma.
Ganglioneuroma:
A fully mature form of the tumor, consisting of benign ganglion cells.
Not cancerous and often found incidentally.
Genetic and Molecular Subtypes
MYCN-Amplified Neuroblastoma:
High levels of MYCN gene amplification are associated with aggressive disease and poor prognosis.
ALK-Mutated Neuroblastoma:
Mutations in the ALK gene can drive neuroblastoma in some cases.
Targeted therapies may be effective.
Non-MYCN-Amplified Neuroblastoma:
Generally associated with a less aggressive disease course.
Risk-Based Types (Stratified by International Neuroblastoma Risk Group)
Low-Risk Neuroblastoma:
Often localized.
Good prognosis with minimal treatment.
Intermediate-Risk Neuroblastoma:
May require a combination of surgery, chemotherapy, and observation.
Prognosis is generally favorable.
High-Risk Neuroblastoma:
Includes cases with metastasis, MYCN amplification, or unfavorable histology.
Requires intensive multimodal treatment (chemotherapy, surgery, stem cell transplant, immunotherapy).
Prognosis can be challenging.
This classification is essential for developing personalized treatment plans and predicting outcomes for children diagnosed with neuroblastoma.
CAUSES AND RISK FACTORS
The exact cause of neuroblastoma is not fully understood, but it arises from genetic and developmental abnormalities in immature nerve cells (neuroblasts). These cells typically mature into normal nerve cells, but in neuroblastoma, they continue to grow uncontrollably. Several factors may contribute to the development of neuroblastoma, although most cases are sporadic.
Causes
Genetic Mutations:
Somatic (non-inherited) mutations in nerve cell precursors during fetal development.
Mutations in specific genes such as ALK (Anaplastic Lymphoma Kinase) and PHOX2B, which play a role in nerve development, can lead to neuroblastoma.
MYCN Amplification:
Some neuroblastomas have amplification of the MYCN oncogene, which is associated with more aggressive tumor growth and poorer outcomes.
Abnormal Neural Crest Development:
Neuroblastoma originates from neural crest cells, which are involved in the development of the nervous system and adrenal glands. Errors in their maturation may lead to neuroblastoma.
Risk Factors
Age:
Neuroblastoma is most common in children under 5 years old.
The majority of cases are diagnosed before the age of 2.
Family History:
A small percentage (1–2%) of cases are familial, suggesting a genetic predisposition.
Familial neuroblastoma is often linked to mutations in the ALK or PHOX2B genes.
Gender:
Slightly more common in boys than in girls.
Environmental Factors:
No specific environmental exposures have been definitively linked to neuroblastoma.
No evidence suggests parental lifestyle or exposure to certain substances during pregnancy is a significant factor.
Congenital Syndromes and Genetic Conditions:
Children with certain genetic syndromes or abnormalities may have an increased risk of neuroblastoma:
Beckwith-Wiedemann Syndrome
Li-Fraumeni Syndrome
Neurofibromatosis Type 1
Hirschsprung Disease
Prenatal Factors:
Errors in fetal development of nerve cells can increase the likelihood of neuroblastoma, although specific triggers are unclear.
Is Neuroblastoma Inherited?
Most cases of neuroblastoma are not inherited and occur sporadically due to mutations in nerve cells.
In the rare familial cases, inheritance is typically autosomal dominant, meaning one copy of the mutated gene can increase the risk, though the likelihood of developing the disease is not 100%.
Understanding these causes and risk factors is essential for developing potential preventive and therapeutic strategies, though currently, there are no known preventive measures for neuroblastoma.
TREATMENT OF NEUROBLASTOMA
The treatment of neuroblastoma depends on the patient’s age, risk group (low, intermediate, or high), tumor location, and disease stage. A multidisciplinary approach is typically used, combining surgery, chemotherapy, radiation therapy, and, in some cases, advanced treatments like immunotherapy or stem cell transplantation.
Low-Risk Neuroblastoma
Treatment:
Observation: Some small, asymptomatic tumors may regress without treatment.
Surgery: Removal of the tumor is often sufficient.
Chemotherapy is rarely needed unless surgery cannot fully remove the tumor or there are complications.
Prognosis:
Excellent, with a high likelihood of cure.
Intermediate-Risk Neuroblastoma
Treatment:
Chemotherapy: Used to shrink the tumor before or after surgery.
Surgery: Performed to remove as much of the tumor as possible.
Radiation therapy is rarely required but may be used if the tumor cannot be completely removed or chemotherapy is insufficient.
Prognosis:
Very good, with effective treatment leading to high survival rates.
High-Risk Neuroblastoma
Treatment:
Intensive Chemotherapy: High doses are used to shrink the tumor and eliminate cancer cells.
Surgery: Aims to remove as much of the tumor as possible.
Stem Cell Transplantation: High-dose chemotherapy followed by autologous stem cell rescue to restore bone marrow function.
Radiation Therapy: Targets the primary tumor site and areas of metastasis.
Immunotherapy:
Anti-GD2 monoclonal antibodies (e.g., dinutuximab) are used to target neuroblastoma cells.
Often combined with cytokines like IL-2 to boost the immune system.
Retinoid Therapy:
13-cis-retinoic acid is used after treatment to promote differentiation of neuroblastoma cells and reduce relapse risk.
Prognosis:
Challenging, but advances in treatment are improving outcomes.
Additional Treatment Strategies
Radiation Therapy:
For tumors that are inoperable or resistant to chemotherapy.
May include proton beam therapy for precise targeting.
MIBG Therapy:
A radioactive isotope (I-131) linked to a molecule called MIBG targets neuroblastoma cells.
Used in high-risk or relapsed cases.
Targeted Therapy:
ALK inhibitors (e.g., crizotinib) for tumors with ALK mutations.
Clinical Trials:
New therapies, including CAR-T cell therapy and novel drugs, are being tested in clinical trials.
Supportive Care
Pain Management: Medications for pain relief.
Nutritional Support: Ensuring adequate nutrition during intensive treatment.
Psychological Support: Counseling for the child and family to cope with the emotional impact of the disease.
Follow-Up and Monitoring
After treatment, regular follow-up visits are essential to monitor for recurrence or long-term side effects of therapy.
Imaging studies, urine tests, and blood work are often part of ongoing care.
The treatment approach is highly personalized, and advances in research continue to improve survival rates and quality of life for children with neuroblastoma.
PREVENTION OF NEUROBLASTOMA
Currently, there is no known way to prevent neuroblastoma, as the disease is primarily caused by genetic mutations and developmental abnormalities in immature nerve cells (neuroblasts), which are not influenced by modifiable lifestyle or environmental factors. Most cases of neuroblastoma are sporadic, meaning they occur without a known inherited or environmental cause.
Why Prevention Is Difficult
Unknown Environmental Factors:
Unlike some cancers, no specific environmental exposures or parental behaviors have been definitively linked to the development of neuroblastoma.
Genetic Origins:
Neuroblastoma often results from spontaneous genetic mutations in early fetal development, which are not influenced by external factors.
Hereditary Cases:
A very small percentage (1–2%) of cases are familial, caused by inherited mutations in genes like ALK or PHOX2B. However, these are rare, and genetic testing may identify families at risk.
What Can Be Done?
Genetic Counseling
Families with a history of neuroblastoma or known genetic mutations can benefit from genetic counseling.
Testing for mutations like ALK or PHOX2B may help assess the risk for future children.
Prenatal and Early Life Care
While no specific measures can prevent neuroblastoma, ensuring general prenatal health through proper nutrition, avoiding harmful substances (like tobacco or alcohol), and regular medical check-ups is recommended.
Early Detection
While not prevention, early detection can improve outcomes. Awareness of symptoms such as persistent pain, lumps, or unusual bruising in young children can prompt earlier medical evaluation.
Research Participation
Families with a history of neuroblastoma may consider participating in research studies aimed at understanding the genetic and biological causes of the disease.
Future Directions
Ongoing research into the genetic and molecular causes of neuroblastoma may eventually lead to prevention strategies, such as:
Identifying and correcting genetic abnormalities early in life.
Developing screening tools for at-risk populations (e.g., familial cases).
At present, the focus remains on improving early diagnosis, treatment, and long-term outcomes rather than prevention.
NEWER ADVANCEMENTS AND RESEARCHES IN NEUROBLASTOMA
Recent advancements in neuroblastoma research have led to significant improvements in understanding the disease and developing innovative treatment strategies. Key areas of progress include:
Immunotherapy Enhancements
DLK1-Directed Immunotherapy: Researchers at the Children’s Hospital of Philadelphia have identified DLK1 as a promising target for immunotherapy in neuroblastoma. Preclinical studies have shown that targeting DLK1 can effectively eliminate neuroblastoma cells, leading to the initiation of a first-in-human clinical trial for this approach.
Targeted Molecular Therapies
ALK Inhibitors: Mutations in the ALK gene are implicated in some neuroblastoma cases. Targeted therapies using ALK inhibitors, such as crizotinib, have shown efficacy in treating tumors with these mutations, offering a more personalized treatment approach.
Radiopharmaceutical Treatments
177Lu-DOTATATE Therapy: Neuroblastoma cells often express somatostatin receptors, making them suitable targets for radiopharmaceutical treatments like 177Lu-DOTATATE. Clinical trials have demonstrated promising results, with this therapy effectively targeting and destroying neuroblastoma cells while sparing healthy tissue.
Genetic and Molecular Research
MYCN Amplification Studies: Amplification of the MYCN gene is associated with high-risk neuroblastoma. Recent research has focused on understanding the role of MYCN in tumor development and progression, leading to potential therapeutic targets aimed at inhibiting its activity.
Clinical Trials and Treatment Protocols
ANR 2025 Conference: The upcoming Advances in Neuroblastoma Research (ANR) meeting in May 2025 in Washington, D.C., will serve as a platform for researchers and clinicians to share the latest findings and treatment strategies, fostering collaboration and innovation in the field.
These advancements represent a concerted effort to improve outcomes for children diagnosed with neuroblastoma, focusing on personalized and targeted therapies that minimize side effects and enhance survival rates.